Evaluates sequences quality
Source:R/AllGenerics.R, R/class-BarcodeQc.R, R/method-seqQc.R
bc_seq_qc.Rdbc_seq_qc evaluates sequences quality. See the return value for detail.
bc_seq_qc(x, sample_name = NULL, reads_sample_size = 1e+05)
bc_plot_seqQc(x)
# S4 method for class 'ShortReadQ'
bc_seq_qc(x, reads_sample_size = 1e+05)
# S4 method for class 'DNAStringSet'
bc_seq_qc(x, reads_sample_size = 1e+05)
# S4 method for class 'data.frame'
bc_seq_qc(x, reads_sample_size = 1e+05)
# S4 method for class 'integer'
bc_seq_qc(x, reads_sample_size = 1e+05)
# S4 method for class 'character'
bc_seq_qc(x, sample_name = basename(x), reads_sample_size = 1e+05)
# S4 method for class 'list'
bc_seq_qc(x, sample_name = names(x))
# S4 method for class 'BarcodeQc'
bc_plot_seqQc(x)
# S4 method for class 'BarcodeQcSet'
bc_plot_seqQc(x)Arguments
- x
A single or list of Fastq files, ShortReadQ object, DNAStringSet object, data.frame or named integer vector.
- sample_name
A character vector with the length of sample number, used to set the sample name.
- reads_sample_size
A integer value defines the sample size of the sequences for quality control analysis. If there are fewer sequences comparing to this value, all the sequences will be used. The default is 1e5.
Value
A barcodeQc or a barcodeQcSet class. The barcodeQc is a list with four slots,
top: adata.framewith top 50 most frequency sequence,distribution: adata.framewith the distribution of read depth. It containsnOccurrences(depth), andnReads(unique sequence) columns.base_quality_per_cycle:data.framewith base-pair location (NGS sequencing cycle) by row, and the base-pair quality summary by column, including Mean, P5 (5 P75 (75base_freq_per_cycle:data.framewith three columns: 1.Cycle, the sequence base-pair location (NGS sequencing cycle); 2.Base, DNA base;Count: reads count.summary: a numeric vector with following elements:
total_read,median_read_length,p5_read_length,p95_read_length.
The barcodeQcSet is a list of barcodeQc.
Examples
library(ShortRead)
# fastq file
fq_file <- system.file("extdata", "simple.fq", package="CellBarcode")
bc_seq_qc(fq_file)
#> Sequnece QC, summary:
#> total_read: 1
#> p5_read_length: 12
#> median_read_length: 12
#> p95_read_length: 12
# ShortReadQ
sr <- readFastq(fq_file[1])
bc_seq_qc(sr)
#> Sequnece QC, summary:
#> total_read: 1
#> p5_read_length: 12
#> median_read_length: 12
#> p95_read_length: 12
# DNAStringSet
ds <- sread(sr)
bc_seq_qc(ds)
#> Sequnece QC, summary:
#> total_read: 1
#> p5_read_length: 12
#> median_read_length: 12
#> p95_read_length: 12
# List of DNAStringSet
l <- list(sample1 = ds, sample2 = ds)
bc_plot_seqQc(bc_seq_qc(l))
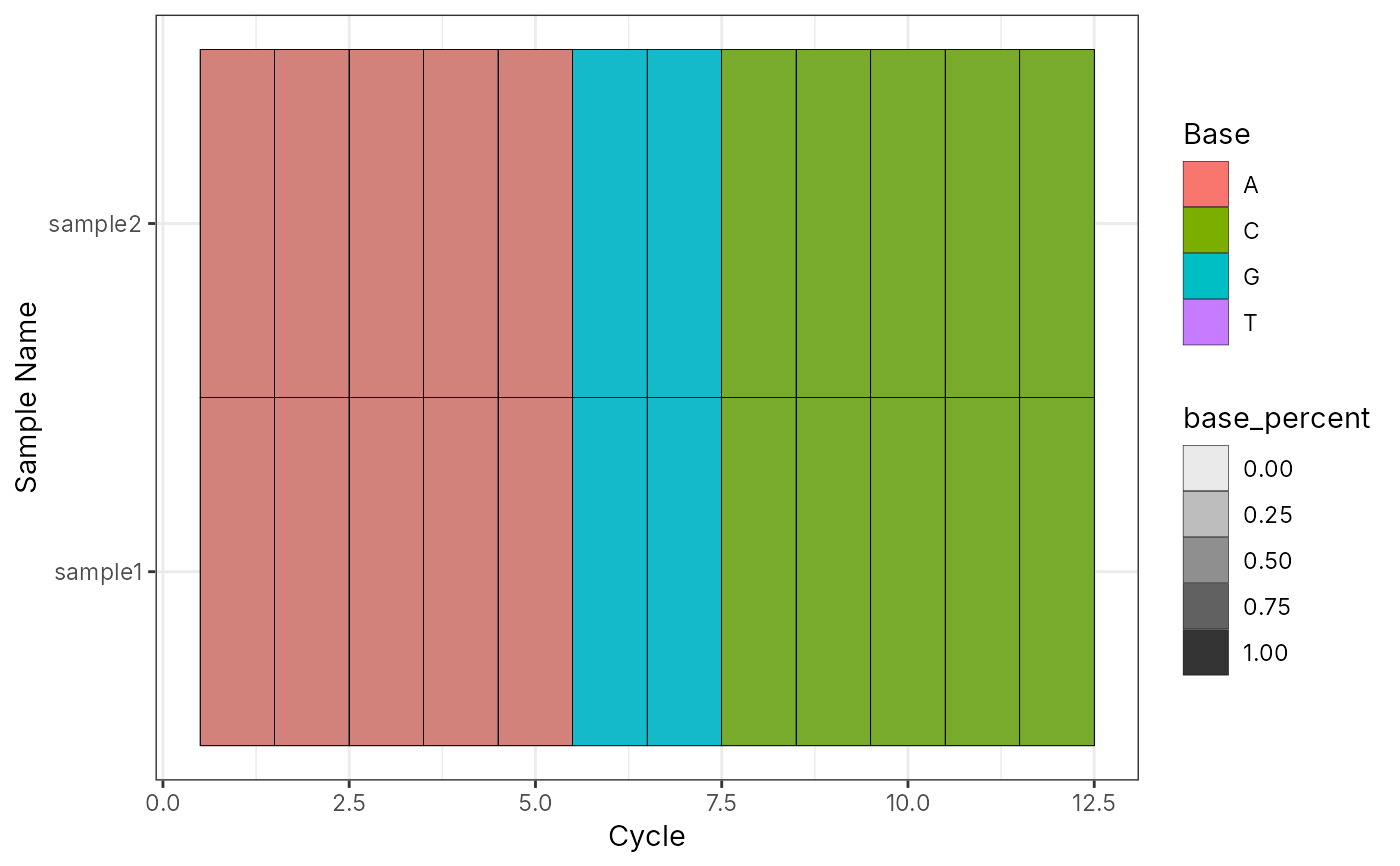 # List of ShortRead
l_sr <- list(sample1 = sr, sample2 = sr)
bc_plot_seqQc(bc_seq_qc(l_sr))
# List of ShortRead
l_sr <- list(sample1 = sr, sample2 = sr)
bc_plot_seqQc(bc_seq_qc(l_sr))
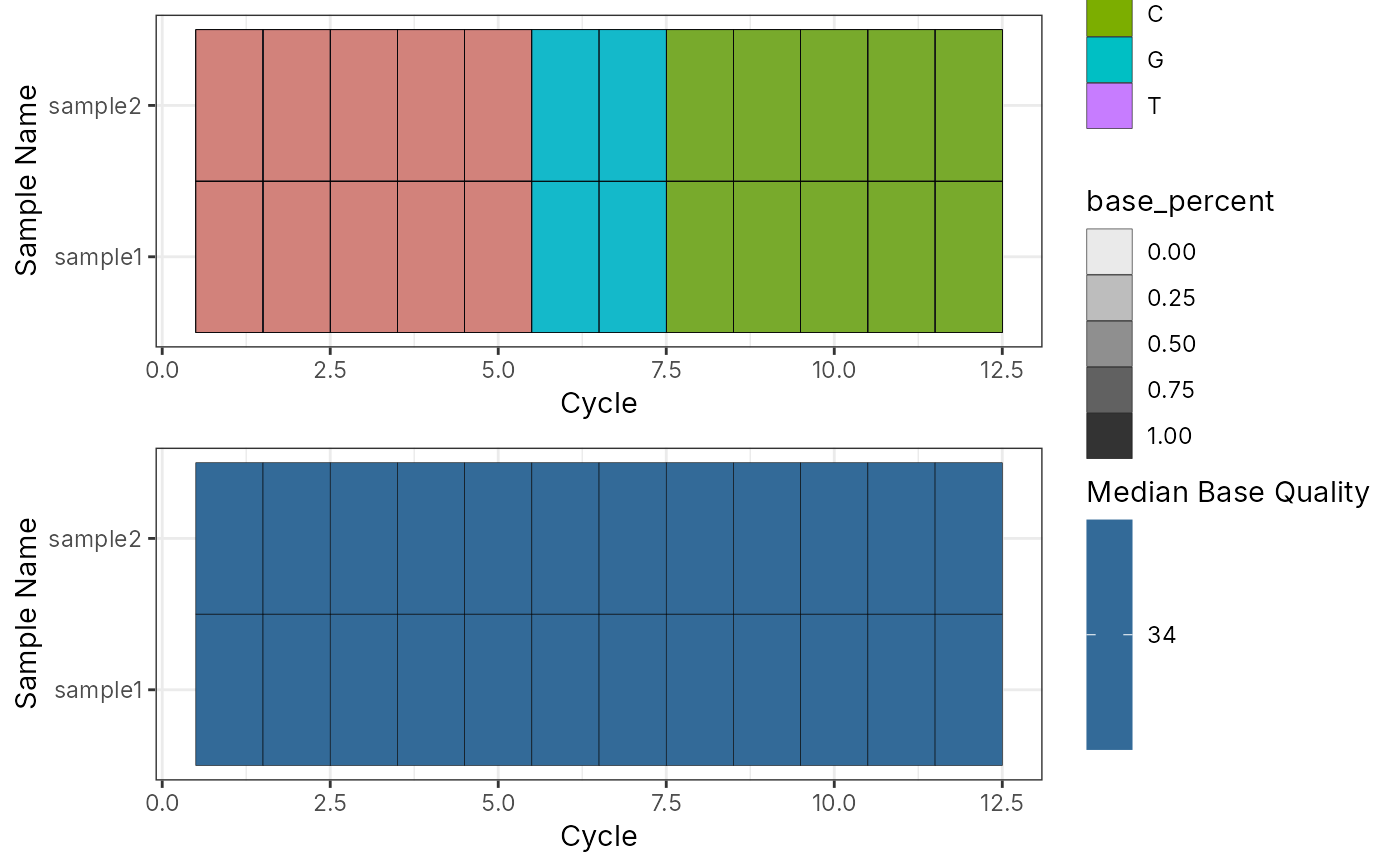 ###
###